
Jane Chen1; Juliana Maria Ferraz Sallum2; Silvia Bragagnolo Longhitano3; Décio Brunoni4
DOI: 10.1590/S0004-27492000000600007
RESUMO
Introdução: Para se verificar a prevalência de anomalias oculares em indivíduos portadores de deficiência auditiva de causa genética definitiva ou suspeita, este trabalho apresenta a avaliação oftalmológica de 97 indivíduos portadores de deficiência auditiva. Pacientes e Métodos: 97 indivíduos com diagnóstico definitivo ou suspeito de causa genética para disacusia foram submetidos a exame clínico oftalmológico completo; destes, 10 foram excluídos. Resultados: 42 (48,28%) dos pacientes apresentaram uma ou mais anomalias oculares, 22 (25,29%) pacientes apresentaram várias anormalidades oculares e quadro clínico compatíveis com síndromes genéticas estabelecidas. Conclusões: O exame oftalmológico é importante no diagnóstico sindrômico e etiológico de alguns quadros de disacusia, pois as alterações oculares podem ser a única anomalia associada à mesma.
Descritores: Anomalias oculares; Deficiência auditiva genética
ABSTRACT
Purpose: In order to verify the prevalence of ocular abnormalities in patients who are deaf due to genetic causes, this paper presents the visual assessment of 97 deaf patients. Methods: 97 patients with definite or suspected diagno- sis of congenital and genetic deafness underwent a complete ophthalmologic evaluation; 10 patients were excluded. Results: 42 (48.28%) patients presented one or more ocular abnormalities, 22 (25.29%) patients presented several abnormalities and clinical manifestations of established genetic diseases. Conclusions: The ocular abnormalities may be the only clinical manifestations associated with deafness. Therefore the ophthalmological examination is a helpful tool for the etiological diagnosis of deafness.
Keywords: Ocular abnormalities; Genetical deafness
INTRODUÇÃO
A frequência de anormalidades oculares encontradas entre indivíduos portadores de deficiência auditiva é mais alta do que nos indivíduos com audição normal 1. No estudo deRegenbogen & Godel (1985) foram examinadas 150 crianças portadoras de deficiência auditiva e 45,3% delas apresentaram anormalidades oculares que levavam a diminuição da visão.
Johnson (1991) também demonstrou que um grande número de pessoas portadoras de deficiência auditiva tinham problemas visuais concomitantes 2. Neste estudo 1435 estudantes que ingressaram na NTID (National Technical Institute for the Deaf) entre os anos de 1987 e 1991, foram examinados e a incidência de problemas visuais encontrada foi de aproximadamente 55%.
Elango, et al (1994) examinou 165 crianças de uma escola para deficientes auditivos na Malásia e encontrou uma prevalência de anomalias oculares de 57,6% 3. Comparativamente, maior que a incidência de problemas visuais encontrados na população geral, descrita em um estudo feito pela NSPB (National Society to Prevent Blindness) com crianças em idade escolar, foi de 25% 4.
OBJETIVOS
Os objetivos deste estudo são verificar a prevalência de anormalidades oculares em indivíduos portadores de deficiência auditiva de causa genética, identificar as principais doenças que apresentam deficiências auditiva e visual associadas e demonstrar a importância da realização de avaliação oftalmológica nestes indivíduos.
PACIENTES E MÉTODOS
Foi realizado um estudo prospectivo no Departamento de Oftalmologia da Universidade Federal de São Paulo - Escola Paulista de Medicina entre os anos de 1992 e 1994. A idade dos pacientes variou de recém-nascidos a 58 anos.
Foram examinados 97 indivíduos encaminhados pela Disciplina de Genética do Departamento de Morfologia da Universidade Federal de São Paulo - Escola Paulista de Medicina para investigação de possível causa genética para a surdez congênita.
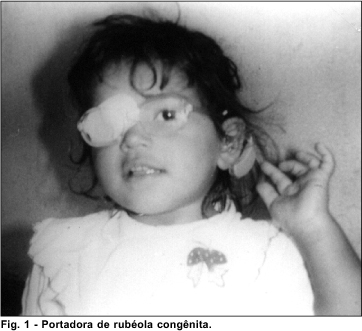
Destes 97 indivíduos, 10 foram excluídos graças à avaliação oftalmológica, que possibilitou identificar vários quadros de infecção congênita nos quais não se supôs este diagnóstico etiológico por ausência de história ou outras alterações (9 casos de rubéola e 1 caso de toxoplasmose).
O exame genético clínico detalhado dos pacientes e de suas famílias foi tema de tese na Disciplina de Genética do Departamento de Morfologia da Universidade Federal de São Paulo - Escola Paulista de Medicina por Longhitano 5. Este exame consta da propedêutica genético-clínica com anamnese e exame físico completos e medidas antropométricas específicas, incluindo verificação do desenvolvimento neuropsicomotor. Os exames complementares realizados foram: avaliação otoscópica direta, estudos audiométricos (audiometria e impedânciometria nos pacientes passíveis de condicionamento ou exame objetivo, audiometria de tronco cerebral ou eletrococleografia, nos pacientes que não condicionavam à audiometria), e tomografia computadorizada do osso temporal, em todos os pacientes. Em alguns pacientes foram realizados outros exames e avaliações complementares (por exemplo: radiografia de tórax, urinálise, eletrocardiograma, eletroencefalograma), conforme a necessidade do caso.
Foi realizado exame oftalmológico completo de cada paciente no Departamento de Oftalmologia da Universidade Federal de São Paulo - Escola Paulista de Medicina.
O exame oftalmológico foi composto de: história, medida da acuidade visual, refração, avaliação da musculatura ocular extrínseca, biomicroscopia, tonometria, quando possível, e oftalmoscopia binocular indireta sob midríase. E, em alguns casos, foram realizados propedêutica de vias lacrimais, nos casos de obstrução das vias lacrimais, estudos eletrofisiológicos, nos casos de Síndrome de Usher e Doença de Norrie, e de visão de cores, nos casos de Síndrome de Usher e atrofia de papila.
RESULTADOS
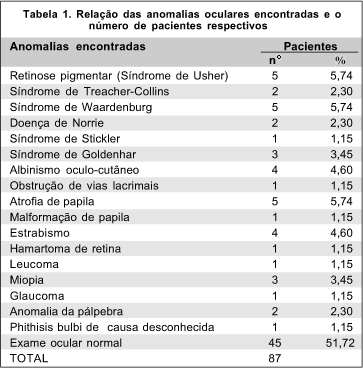
Quarenta e dois pacientes (48,28%) dos oitenta e sete examinados apresentaram uma ou mais anormalidades oculares (tabela I). Vinte e dois pacientes (25,29%) apresentaram várias anormalidades oculares e quadro clínico compatíveis com síndromes genéticas estabelecidas.
DISCUSSÃO
No presente trabalho, o exame clínico oftalmológico completo revelou alterações oculares em cerca de metade dos pacientes. Dos 87 pacientes examinados, 5 (5,74%) com Síndrome de Usher, 2 (2,30%) com Síndrome de Treacher-Collins, 5 (5,74%) com Síndrome de Waardenburg, 2 (2,30%) com Doença de Norrie, 1 (1,15%) com Síndrome de Stickler, 3 (3,45%) com Síndrome de Goldenhar e 4 (4,60%) com Albinismo oculo-cutâneo.
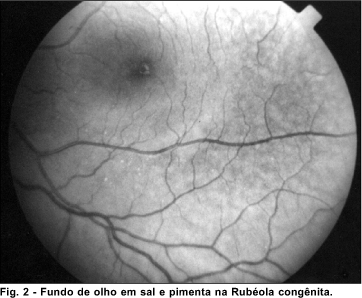
Dos pacientes que foram excluídos deste trabalho, nove apresentaram a retina com aspecto em sal e pimenta, o que nos leva ao diagnóstico de infecção congênita por rubéola, permitindo excluir o diagnóstico de deficiência auditiva congênita de causa genética.
A retinopatia pigmentar ou em sal e pimenta é a lesão ocular mais comumente encontrada relacionada à rubéola congênita e é caracterizada por áreas de hiperpigmentação e áreas de hipopigmentação do epitélio pigmentário da retina. Outras lesões oculares que podem estar associadas são: catarata, glaucoma, microftalmia, iridociclite, nistagmo, alta miopia e estrabismo.
Outra infecção congênita encontrada em um dos pacientes foi a toxoplasmose congênita. Estes casos apresentam cicatriz de coriorretinite, em geral macular, que causa baixa acentuada de acuidade visual.
Não foi realizada uma abordagem sorológica sistemática, uma vez que os testes sorológicos só apresentam valor diagnóstico se realizados precocemente ou para exclusão do diagnóstico. A identificação dos quadros infecciosos foi feita através das alterações encontradas na fundoscopia.
Síndrome de Usher
A Síndrome de Usher 8 (OMIM *276900) é uma condição genética com heterogeneidade não alélica e formas clínicas distintas.
Na Síndrome de Usher a perda auditiva neurossensorial hereditária em graus variados está associada a retinose pigmentária (RP). A RP é uma degeneração hereditária e progressiva da retina. Fotorreceptores, células ciliadas auditivas e células ciliadas vestibulares se desenvolvem a partir de células ciliadas progenitoras. Inúmeras evidências sugerem que uma anormalidade generalizada de estruturas do axonema está presente em pacientes com Síndrome de Usher.
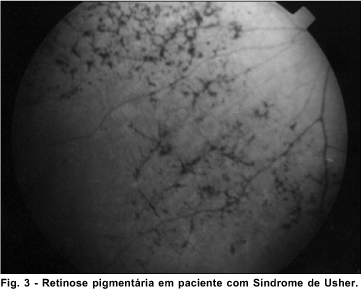
A síndrome é classificada nos seguintes tipos:
- tipo I: Condição autossômica recessiva com heterogeneidade genética. Caracterizada por surdez congênita severa, instalação precoce de RP e ausência de funções vestibulares. Apresenta os seguintes subtipos, classificados de acordo com o locus gênico:
1A(OMIM*276900) - 14q 32;
1B(OMIM*276903) - 11q;
1C(OMIM*276904) - 11p15.1;
1D(OMIM*601067) - 10q;
1E(OMIM*602097) - 21q21;
1F(OMIM*602083) - Chr.10.
- tipo II: Condição autossômica recessiva com heterogeneidade genética. Caracterizada por surdez congênita moderada a severa, instalação tardia de RP e funções vestibulares normais. Apresenta dois subtipos:
2A(OMIM*276901) - 1q32-q41;
2B(OMIM*276905) - o lócus ainda não foi definido
-tipo III (OMIM*276902): Condição autossômica recessiva, lócus mapeado em 3q21-q25. Caracterizada por visão e audição inicialmente normais ou próximas do normal que pioram progressivamente ao longo de décadas.
- tipo IV: forma ligada ao X.
Outras manifestações clínicas associadas podem ser: perda visual progressiva "tipo retinose pigmentária", catarata, anormalidades vestibulares, retardo mental, ataxia e psicose.
Nos 5 pacientes examinados foi encontrada a retinose pigmentária.
Síndrome de Treacher-Collins
A Síndrome de Treacher-Collins 8 (Disostose mandibulofacial)(OMIM*154500) apresenta herança autossômica dominante e é uma entidade clinicamente heterogênea. O lócus desta doença foi mapeado em 5q32-33.1 e existem evidências de que a idade paterna influencia na ocorrência de mutações novas.
As manifestações clínicas dessa doença são: fenda palpebral antimongolóide, coloboma de pálpebra inferior, micrognatia, microtia, deformidade da orelha, hipoplasia dos arcos zigomáticos e macrostomia. O diagnóstico diferencial se faz com a Síndrome de Nager (disostose acrofacial) e com a Síndrome de Goldenhar (espectro oculoauriculovertebral).
Nos dois pacientes examinados foram encontrados todas as características oculares.
Síndrome de Waardenburg
A Síndrome de Waardenburg 8 (OMIM*193500) apresenta herança autossômica dominante com penetrância incompleta e expressividade variável 6. Existem evidências de que a idade paterna influencia na ocorrência de mutações novas.
Existem quatro subtipos moleculares desta síndrome, que são baseados em uma heterogeneidade genética:
- tipo I (OMIM*193500): Todos os subtipos do tipo I apresentam mutação do gene PAX3 no cromossomo 2q35;
- tipo II (OMIM#193510): Mutação no gene MITF. Apresenta dois subtipos:
IIA - Mutação em um gene no
cromossomo 3p12.3-14.1;IIB - Mutação em um gene no
cromossomo 1p21-p13.3;
- tipo III (OMIM#148820): Trata-se também de uma mutação no gene PAX3, podendo ser considerado uma variação de expressão extrema do tipo I, ou de uma síndrome do gene contíguo devido à deleção do gene PAX3 e de genes adjacentes.
- tipo IV (OMIM#277580): Mutação em 22q13 (gene SOX10), 20q13.2-q13.3 (geneEDN3)e 13q22 (gene EDNRB).
Em todos os quatro tipos ocorre perda auditiva neurossensorial. A surdez é secundária à disgenesia coclear, uni ou bilateral e de diferentes graus de acometimento. As outras manifestações clínicas dos subtipos desta síndrome são: deslocamento lateral do canto medial e do ponto lacrimal inferior (distopia, encontrado no tipo I), base do nariz alargada, heterocromia total ou parcial da íris, fundo de olho albinóide, hiperplasia da porção medial dos supercílios frequentemente com sinofris superciliar, mecha branca frontal no cabelo ou canície precoce, associação com fendas palatinas e labiais, nariz com abas pequenas, narinas fechadas e extremidade arredondada e assimetria facial.
Na heterocromia da íris, a íris hipopigmentada apresenta uma coloração azul clara, pálida por hipoplasia do estroma, que é visível à biomicroscopia.
Os 5 pacientes com Síndrome de Waardenburg examinados neste trabalho apresentavam todas as características oculares. Exceto um paciente que não apresentava fundo de olho totalmente albino, era apenas hipopigmentado em algumas áreas da retina.
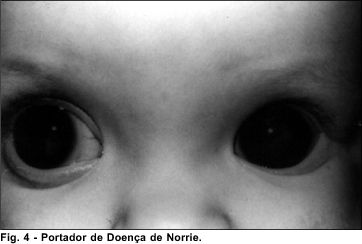
Doença de Norrie
A Doença de Norrie 8 (OMIM*310600) apresenta herança ligada ao X recessiva e foi mapeada para o locus Xp11.4.
As manifestações clínicas descritas nesta doença são: perda auditiva neurossensorial progressiva, retardo mental e alterações oculares. As manifestações oculares descritas são: pseudotumor de retina bilateral e congênito, hiperplasia de retina, displasia retiniana em geral grave, hipoplasia e necrose das camadas mais internas da retina, catarata, phthisis bulbi e pode ter descolamento de retina.
A Doença de Norrie é frequentemente de difícil diagnóstico porque mais da metade dos pacientes não apresenta nem perda auditiva e nem retardo mental, consideradas, junto aos sintomas oculares específicos, características clássicas.
Nos 2 pacientes examinados foram encontradas a displasia retiniana severa.
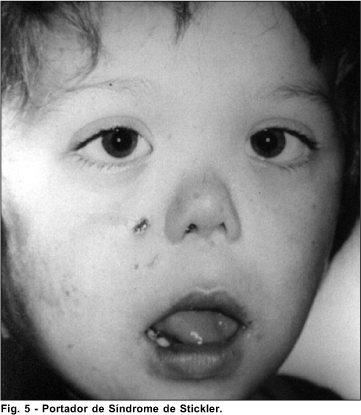
Síndrome de Stickler
A Síndrome de Stickler 8 (Artro-oftalmopatia hereditária)(OMIM#108300 e #184840), inicialmente descrita em 1965, é de herança autossômica dominante, trata-se de uma desordem do tecido conjuntivo. A sua prevalência é estimada em 1:10000 pessoas 7.
A Síndrome de Stickler é classificada em três tipos, de acordo com o gene no qual ocorre a mutação: tipo I por mutação no gene do COL2A1 (OMIM#108300), tipo II por mutação no gene do COL11A2 (OMIM#184840) e tipo III por mutação no gene do COL11A1 (OMIM#121028). Existem evidências que sugerem a existência de um quarto tipo desta síndrome.
As manifestações clínicas encontradas nesta síndrome são anormalidades oculares e orofaciais, artropatia e deficiência auditiva. As manifestações oculares, descritas podem ser: miopia de moderada a severa com início na primeira década da vida, descolamento de retina, degeneração vitreorretiniana, catarata e glaucoma. As alterações orofaciais descritas são: achatamento da região média da face, complexo de Pierre Robin (micrognatia, glossoptose e anormalidade da mobilidade do palato). Podem ser encontradas também estatura baixa ou fenótipo marfanóide com hiperextensibilidade articular e artrite precoce. Aproximadamente 50% dos pacientes podem apresentar prolapso de valva mitral. A surdez é geralmente neurossensorial, podendo ter também um componente condutivo.
Nos 2 pacientes examinados formam encontrados todas as alterações oculares. Um deles apresentou além do quadro referido, palidez de papila.
Síndrome de Goldenhar
Na Síndrome de Goldenhar 8 (Espectro oculoauriculovertebral)(OMIM164210) muitos casos são esporádicos, algumas famílias apresentaram herança autossômica recessiva, outras apresentaram herança claramente autossômica dominante. É uma entidade geneticamente heterogênea. Os defeitos encontrados nesta síndrome são devidos a problemas na morfogênese dos primeiro e segundo arcos branquiais, associados a anomalias vertebrais e/ou oculares.
As características clínicas desta síndrome são: deformidade da orelha externa até anotia, apêndices pré-auriculares geralmente anteriores ao tragus, implantação baixa das orelhas, lipodermóide da conjuntiva ou córnea, coloboma em pálpebra superior, microftalmia ou anoftalmia, anomalias de orelha média levando a perda auditiva de natureza condutiva e anomalias vertebrais.
Nos três pacientes examinados foram encontrados todas as alterações oculares.
Outras malformações oculares isoladas também foram encontradas, como por exemplo: atrofia de papila, malformação de papila, anomalia de pálpebra, hamartoma de retina, leucoma, miopia, glaucoma, albinismo oculo-cutâneo, obstrução das vias lacrimais, phithisis bulbi e estrabismo. Entretanto, não foi possível juntar estes diagnósticos em um diagnóstico sindrômico ou etiológico estabelecido.
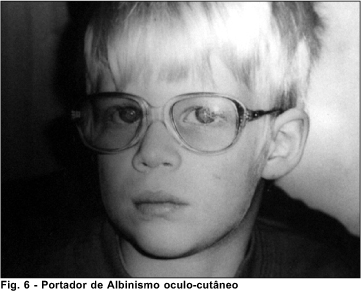
Albinismo oculo-cutâneo
O albinismo óculo-cutâneo associado à deficiência auditiva apresenta herança autossômica recessiva. Isoladamente apresenta heterogeneidade não-alélica e formas clínicas distintas. É caracterizado por diminuição da pigmentação de pele e/ou pêlos e/ou olhos. É classificado nos seguintes tipos:
- tipo I (ou albinismo óculo-cutâneo tirosinase-negativa) 8 (OMIM*203100): Resulta de mutações no gene da tirosinase que levam a perda da função da enzima tirosinase sobre o melanócito. Variam desde ausência total de pigmento até a pigmentação próxima do normal, uma importante característica é a presença de hipopigmentação marcante ao nascimento. Apresenta os subtipos 1A e 1B, ambos mapeados para o lócus 11q14-q21.
- tipo II (ou albinismo óculo-cutâneo tirosinase-positiva) 8 (OMIM*203200): É o tipo mais prevalente de albinismo óculo-cutâneo. É caracterizado pela presença de certa pigmentação ao nascimento e que se reduz com o tempo. Foi mapeado para o lócus 15q11.2-q12.
- tipo III (ou albinismo marrom) 8 (OMIM#203290): Mapeado para o lócus 9p23, caracteriza-se por cabelo de cor castanho claro e íris de cor variando de cinza a castanho ao nascimento. Com o passar do tempo, ocorre apenas discreta alteração na cor da pele, porém, o cabelo e a íris tornam-se mais escuros.
Além destes, diversos quadros de síndromes com graus variáveis de hipopigmentação são descritos na literatura.
Os achados oculares frequentemente encontrados nos vários tipos de albinismo óculo-cutâneo podem ser: diminuição da pigmentação da íris e retina, nistagmo, fotofobia, erros refracionais e diminuição da acuidade visual.
CONCLUSÕES
A prevalência de alterações oculares em pacientes com disacusia foi de 48,28%. Esta prevalência foi semelhante aos encontrados por: Regenbogen & Godel (1985) 1 (45,3%); Johnson (1991) 2 (55%); Elango et al (1994) 3 (57,6%) e pelo estudo da National Society to Prevent Blindness 4 (25%)
As principais doenças que apresentam deficiência auditiva de causa genética e deficiência visual associadas são as síndromes de Usher, de Treacher-Collins, de Waardenburg, de Norrie, de Stickler e de Goldenhar e albinismo oculo-cutâneo.
O exame oftalmológico é importante no diagnóstico sindrômico e etiológico de alguns quadros de disacusia, pois as alterações oculares podem ser a única anomalia associada à mesma 5. É importante também na identificação de casos não-genéticos, como as infecções congênitas por rubéola, na qual o fundo de olho em sal e pimenta pode ser a única anomalia.
Os resultados deste estudo mostram a importância de se estabelecer a avaliação oftalmológica na rotina para investigação de causas genéticas em pacientes portadores de deficiência auditiva.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Regenbogen L, Godel V. Ocular deficiences in deaf children. J Pediatr Ophthalmol Strabismus 1985;22:231-3.
2. Johnson D. Visual assessment of people who are deaf. ASHA (American Speech-Language-Hearing Association), 1991 November.
3. Elango S; Krishna R ; Shriwas SR. Ocular abnormalities in children from a Malaysian school for the deaf. Ann Trop Paediatr 1994;14:149-52.
4. National Society to Prevent Blindness (NSPB). Vision screening of children (p-257). 1980, New York: NSPB.
5. Longhitano SB. Estudo genético-clínico de pacientes portadores de deficiência auditiva genética. Tese apresentada à Universidade Federal de São Paulo - Escola Paulista de Medicina para obtenção de título de Mestre em Morfologia . São Paulo, 1995.
6. Read AP; Newton VE. Waardenburg syndrome. J Med Genet, 1997;34:656-65.
7. MacDonaldd MR, Kolodziej P, Schaefer GB and Olney AH. Stickler syndrome. ENT-Ear, Nose & Throat Journal, 1997;76(10):706.
8. Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD; World Wide Web URL: http://www.ncbi.nlm.nih.gov/omim/
Apresentado sob a forma de tema livre em: II Congresso da Sociedade Brasileira de Retina e Vítreo em Porto Alegre, RS - Brasil, 20 a 22 de maio/1999; XXX Congresso Brasileiro de Oftalmologia em Recife, PE - Brasil, 4 a 7 de setembro/1999; e X Congresso Acadêmico Paulista de Medicina em São Paulo, SP - Brasil, 6 e 7 de outubro/1999.
(1) Aluna de Medicina da Universidade Federal de São Paulo - Escola Paulista de Medicina.
(2)Médica, Doutora em Oftalmologia pela Universidade Federal de São Paulo - Escola Paulista de Medicina. Chefe do Setor de Retina e Vítreo do Departamento de Oftalmologia da Universidade Federal de São Paulo - Escola Paulista de Medicina. Realizou todos os exames oftalmológicos.
(3) Médica contratada, Mestre em Morfologia pela Universidade Federal de São Paulo - Escola Paulista de Medicina.
(4) Professor Adjunto da Disciplina de Genética do Departamento de Morfologia da Universidade Federal de São Paulo - Escola Paulista de Medicina.
Endereço para correspondência: Dra. Juliana M. Ferraz Sallum - Av. Indianópolis, 1797 - São Paulo (SP) CEP 04063-003. Fone/Fax: (11)5585-1136. E-mail: [email protected]
© 2024 - All rights reserved - Conselho Brasileiro de Oftalmologia
![]()
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Scielo
Scielo
 Pocket
Pocket
