
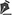
Luis Jesuino de Oliveira Andrade1; Rafael Andrade2; Caroline Santos França3; Alcina Vinhaes Bittencourt4
DOI: 10.1590/S0004-27492009000500019
ABSTRACT
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive disorder with clinical and genetic heterogeneity. This syndrome was first described by Laurence and Moon in 1866 and additional cases were described by Bardet and Biedl between 1920 and 1922. The main features are obesity, polydactyly, pigmentary retinopathy, learning disabilities, various degrees of intellectual impairment, hypogonadism, and renal abnormalities. Bardet-Biedl syndrome is both phenotypically and genetically heterogeneous. Clinical diagnosis is based on the presence of 4 of the 5 cardinal features. The authors present a typical case of pigmentary retinopathy due to Bardet-Biedl syndrome and made a brief commentary about the disease's cardinal manifestations.
Keywords: Bardet-Biedl syndrome; Retinitis pigmentosa; Retinal degeneration; Human; Female; Adolescent; Case reports
RESUMO
A síndrome de Bardet-Biedl (BBS) é uma desordem autossômica recessiva rara, com heterogeneidade clínica e genética. Esta síndrome foi descrita pela primeira vez por Laurence e Moon em 1866 e outros casos foram descritos por Bardet e Biedl entre 1920 e 1922. As principais características são obesidade, polidactilia, retinopatia pigmentar, dificuldades de aprendizagem, graus de deficiência intelectual diversos, hipogonadismo e anomalias renais. Síndrome de Bardet-Biedl é fenotipicamente e geneticamente heterogêneos. O diagnóstico clínico baseia-se na presença de quatro dos cinco sinais principais da síndrome. Os autores apresentam um caso típico de retinopatia pigmentar devido à síndrome de Bardet-Biedl e fazem uma breve revisão sobre as manifestações da síndrome com especial atenção à retinopatia pigmentar.
Descritores: Síndrome de Bardet-Biedl; Retinite pigmentosa; Degeneração retiniana; Humano; Feminino; Adolescente; Relatos de casos
CASE REPORT
Pigmentary retinopathy due to Bardet-Biedl syndrome: case report and literature review
Retinopatia pigmentar devido a síndrome de Bardet-Biedl: relato de caso e revisão da literatura
Luis Jesuino de Oliveira AndradeI; Rafael AndradeII; Caroline Santos FrançaIII; Alcina Vinhaes BittencourtIV
IProfessor Assistente da Faculdade de Medicina da Universidade Estadual de Santa Cruz - UESC - Ilhéus (BA) - Brazil
IIDoutor pela Universidade Federal de São Paulo - UNIFESP - São Paulo (SP)
IIIMédica Residente do Serviço de Oftalmologia do Hospital Universitário Prof. Edgard Santos - Universidade Federal da Bahia - UFBA - Salvador (BA) - Brazil
IVProfessora Adjunta da UFBA - Salvador (BA) - Brazil
ABSTRACT
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive disorder with clinical and genetic heterogeneity. This syndrome was first described by Laurence and Moon in 1866 and additional cases were described by Bardet and Biedl between 1920 and 1922. The main features are obesity, polydactyly, pigmentary retinopathy, learning disabilities, various degrees of intellectual impairment, hypogonadism, and renal abnormalities. Bardet-Biedl syndrome is both phenotypically and genetically heterogeneous. Clinical diagnosis is based on the presence of 4 of the 5 cardinal features. The authors present a typical case of pigmentary retinopathy due to Bardet-Biedl syndrome and made a brief commentary about the disease's cardinal manifestations.
Keywords: Bardet-Biedl syndrome; Retinitis pigmentosa; Retinal degeneration; Human; Female; Adolescent; Case reports; [Publication type]
RESUMO
A síndrome de Bardet-Biedl (BBS) é uma desordem autossômica recessiva rara, com heterogeneidade clínica e genética. Esta síndrome foi descrita pela primeira vez por Laurence e Moon em 1866 e outros casos foram descritos por Bardet e Biedl entre 1920 e 1922. As principais características são obesidade, polidactilia, retinopatia pigmentar, dificuldades de aprendizagem, graus de deficiência intelectual diversos, hipogonadismo e anomalias renais. Síndrome de Bardet-Biedl é fenotipicamente e geneticamente heterogêneos. O diagnóstico clínico baseia-se na presença de quatro dos cinco sinais principais da síndrome. Os autores apresentam um caso típico de retinopatia pigmentar devido à síndrome de Bardet-Biedl e fazem uma breve revisão sobre as manifestações da síndrome com especial atenção à retinopatia pigmentar.
Descritores: Síndrome de Bardet-Biedl; Retinite pigmentosa; Degeneração retiniana; Humano; Feminino; Adolescente; Relatos de casos [Tipo de publicação]
INTRODUCTION
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive disorder with clinical and genetic heterogeneity. The main features are obesity, polydactyly, pigmentary retinopathy, learning disabilities, various degrees of intellectual impairment, hypogonadism, and renal abnormalities. Heterozygous carriers of a BBS mutation are not thought to present symptoms of BBS; however, a previous study reported an increased risk of renal cancer among relatives of patients with BBS. Other clinical features include speech disorder, brachydactyly, developmental delay, polyuria and polydipsia, ataxia, poor coordination/clumsiness, diabetes mellitus, left ventricular hypertrophy, hepatic fibrosis, and renal hypoplasia/dysplasia. Bardet-Biedl syndrome is both phenotypically and genetically heterogeneous. Clinical diagnosis is based on the presence of 4 of the 5 cardinal features(1).
Dental anomalies, regarded as secondary manifestations, include hypodontia, microdontia, short roots, and deep palate. Few reports in the literature have described the oral manifestations of BBS(2). If spasticity and mental retardation are present, it fulfills the criteria for Laurence-Moon-Bardet-Biedl syndrome(3).
The diagnosis of Bardet-Biedl syndrome is established by the clinical findings. Twelve genes are known to be associated with Bardet-Biedl syndrome: BBS1, BBS2, ARL6/BBS3, BBS4, BBS5, MKKS/BBS6, BBS7, TTC8/BBS8, B1/BBS9, BBS10, TRIM32/BBS11, and BBS12. Molecular genetic testing is available on a clinical basis for p.M390R, the common mutation in BBS1 that is present in approximately 18% - 32% of individuals with BBS and p.C91LfsX4 (also known as C91fsX95), a common mutation in BBS10, that is present in 10% of individuals with BBS. However, despite the identification of 12 BBS genes, the molecular basis of BBS remains elusive, it is now evident that all of the known BBS proteins are components of the centrosome and/or basal body and have an impact on ciliary transport(4).
The most common feature of BBS is retinal dystrophy. The retinal appearance in the condition is quite variable with typical retinitis pigmentosa being present in only a minority of cases. The associated optic atrophy can be primary in nature and might play a role in the decreased central vision. Diagnosis of the condition is important for visual prognosis and low vision management. Ophthalmologic and electrophysiological examinations are essential for confirmation and correct diagnosis of BBS(5).
Retinal dysfunction in BBS is well recognized. However, even in early childhood, analysis of retinal function in BBS is frustrated by the attenuation of electroretinograms responses. This precludes an informative analysis of the physiologic processes in many individuals with BBS. An anatomical study of a BBS retina showed good preservation of the inner retina while the photoreceptor layer was degenerated, suggesting that the photoreceptors are the primary retinal site of BBS(6).
BBS is relatively uncommon in Caucasian populations, its incidence has been estimated to be 1 in 150,000 - 160,000 individuals in North America and European populations, but is much higher in some populations with a high level of consanguinity or that are geographically isolated, although higher incidence has been reported in the isolated populations of Newfoundland 1 in 13,000 and Kuwait 1 in 17,000 live births(7).
The authors present a typical case of pigmentary retinopathy due to Bardet-Biedl syndrome, and made a brief commentary about the disease's cardinal manifestations.
CASE REPORT
The patient was a 14 year-old and 8 months female that was seen in the Eyes Hospital Beira-Rio, in Itabuna-Bahia-Brazil, where the diagnosis of pigmentary retinopathy secondary to Bardet-Biedl syndrome was established. The ophthalmic examination revealed visual acuity of 20/200 in both eyes, with refraction of -9,5 sph -6,0 cyl ax 7º in right eye and -9,25 sph -6,0 cyl ax 170º in left eye. Fundus examination showed normal optic nerve, attenuated retinal vessels and diffuse hypo and hyperpigmentation sparing the macular area, as well as bone spicules in the mid peripheral retina (retinitis pigmentosa) (Figure 1A and 1B).
Parental consanguinity was denied and no relatives were known to have the disease. The birth of the patient had been normal. She had learning disabilities from birth. Physical development was within the normal range of chronological age, but mental development was lagged behind the normal range. The vital signs were within normal limits. Physical examination showed: height 156 cm, weight 77.6 kg, and severe obesity of head, trunk, and limbs.
In the present case the phenotype was typical of BBS, despite the absence of polydactyly. Clinical diagnosis is based on the presence of 4 of the 5 cardinal features.
Retinal dysfunction in BBS - review
Retinitis pigmentosa is the name given to a group of genetic eye diseases that affect the retina, and that cause gradual, permanent loss of some vision. As the disease progresses, the retina develops a distinctive appearance that is caused by pigmentary clumping. A striking feature of eye is the lack of cast vessels in the retinal mid periphery displaying bone spicules characteristic of retinal dystrophy. The bone spicules probably represent melanin from retinal pigment epithelial cells migrated into the inner retina(8).
The pathogenesis of retinal dystrophy in BBS is unknown. Current evidence suggests that the primary event in the pathogenesis of BBS is degenerative loss of retinal photoreceptors, with secondary involvement of the inner retinal layers and the retinal pigment epithelium. This evidence is supported by histopathological studies. The electrophysiological findings of reduced or absent electroretinograms responses from retinal photoreceptors, and frequently occurring electro-oculograms changes indicating damage to the pigment epithelium(9).
The retinal dystrophy varied with respect to both the onset of symptoms and the course of the disease, however, clinically disparate phenotypes within the same family have been observed in patients with retinal dystrophy, who had a deletion in the peripherin/RDS gene.
Visual acuity, central retinal function mediated by cones, dark adaptation, and peripheral visual fields (peripheral retina function mediated by rods) are affected. Optic disks and retinal vessels are normal in infancy; disk pallor and attenuated retinal vessels develop with age. Pigmentary changes are observed in the peripheral fundus. Significant cone-rod dystrophy is not apparent in most children under five years of age and cooperation with electroretinograms testing at that age is often poor. Unless strongly indicated, electroretinograms testing may be deferred until at least four years of age(10).
The visual prognosis for children with Bardet-Biedl syndrome is poor, progressive loss of visual acuity arised early in the first decade of life.
REFERENCES
1. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437-46.
2. Drugowick RM, Da Ros Goncalves L, Barroso AS, Feres-Filho EJ, Maia LC. Treatment of gingival overgrowth in a child with Bardet-Biedl syndrome. J Periodontol. 2007;78(6):1159-63.
3. Iannello S, Bosco P, Cavaleri A, Camuto M, Milazzo P, Belfiore F. A review of the literature of Bardet-Biedl disease and report of three cases associated with metabolic syndrome and diagnosed after the age of fifty. Obes Rev. 2002;3(2): 123-35.
4. Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201-13. Comment in: Cell. 2007;129(6):1041-3.
5. Ingster-Moati I, Rigaudiere F, Choltus-De Petigny MC, Bremond-Gignac D, Lestrade C, Grall Y. [Functional visual explorations of Bardet-Biedl syndrome. A study of three cases]. J Fr Ophtalmol. 2000;23(8):802-8.
6. Spaggiari E, Salati R, Nicolini P, Borgatti R, Pozzoli U, Polenghi F. Evolution of ocular clinical and electrophysiological findings in pediatric Bardet-Biedl syndrome. Int Ophthalmol. 1999;23(2):61-7.
7. Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet. 2005; 132(4):352-60.
8. Bek T, Rosenberg T. Clinical pathology and retinal vascular structure in the Bardet-Biedl syndrome. Br J Ophthalmol. 1995;79(1):76-80.
9. Katsumi O, Tanino T, Hirose T, Larson EW, Skladzien CJ. Laurence-Moon-Bardet-Biedl syndrome: electrophysiological and psychophysical findings. Jpn J Ophthalmol. 1985;29(3):282-9.
10. Riise R, Andréasson S, Wright AF, Tornqvist K. Ocular findings in the Laurence-Moon-Bardet-Biedl syndrome. Acta Ophthalmol Scand. 1996;74(6): 612-7.
 Address for correspondence:
Address for correspondence:
Luis Jesuino de Oliveira Andrade
Rua São Marcelo, 246 - Itabuna (BA)
CEP 45600-700
E-mail: [email protected]
Recebido para publicação em 15.01.2009
Última versão recebida em 21.05.2009
Aprovação em 25.05.2009
This paper was conducted at the Endocrinology Service, Hospital Calixto Midlej Filho and Hospital de Olhos Beira-Rio - Itabuna (BA) - Brazil.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência do Dr. José Carlos Molinari Gomes sobre a divulgação de seu nome como revisor, agradecemos sua participação neste processo.
© 2025 - All rights reserved - Conselho Brasileiro de Oftalmologia
![]()
 English PDF
English PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Scielo
Scielo
 Pocket
Pocket

{kind=link}