
Elvira Barbosa Abreu1; Gustavo Amorin Novaes2; Bruno Franco Fernandes2; Patricia Rusa Pereira Odashiro2; Alexandre Nakao Odashiro2; Isabella de Oliveira Lima Parizotto3; Miguel Noel Burnier Jr.2
DOI: 10.1590/S0004-27492012000600003
ABSTRACT
INTRODUCTION: Corneal dystrophy is defined as bilateral and symmetric primary corneal disease, without previous associated ocular inflammation. Corneal dystrophies are classified according to the involved corneal layer in superficial, stromal, and posterior dystrophy. Incidence of each dystrophy varies according to the geographic region studied. PURPOSE: To evaluate the prevalence of stromal corneal dystrophies among corneal buttons specimens obtained by penetrating keratoplasty (PK) in an ocular pathology laboratory and to correlate the diagnosis with patient age and gender. METHODS: Corneal button cases of penetrating keratoplasty from January-1996 to May-2009 were retrieved from the archives of The Henry C. Witelson Ophthalmic Pathology Laboratory and Registry, Montreal, Canada. The cases with histopathological diagnosis of stromal corneal dystrophies were stained with special stains (Peroxid acid Schiff, Masson trichrome, Congo red analyzed under polarized light, and alcian blue) for classification and correlated with epidemiological information (age at time of PK and gender) from patients' file. RESULTS: 1,300 corneal buttons cases with clinical diagnose of corneal dystrophy were retrieved. Stromal corneal dystrophy was found in 40 (3.1%) cases. Lattice corneal dystrophy was the most prevalent with 26 cases (65%). Nineteen were female (73.07%) and the PK was performed at average age of 59.3 years old. Combined corneal dystrophy was found in 8 (20%) cases, 5 (62.5%) of them were female and the average age of the penetrating keratoplasty was 54.8 years old. Granular corneal dystrophy was represented by 5 (12.5%) cases, and 2 (40%) of them were female. Penetrating keratoplasty was performed at average age of 39.5 years old in granular corneal dystrophy cases. Macular corneal dystrophy was present in only 1 (2.5%) case, in a 36 years old female. CONCLUSION: Systematic histopathological approach and evaluation, including special stains in all stromal corneal dystrophies is critical to establish the correct diagnosis.
Keywords: Corneal dystrophies; hereditary; Corneal dystrophies; hereditary; Keratoplasty; penetrating; Corneal stroma
RESUMO
INTRODUÇÃO: A distrofia corneana é definida como doença primária da córnea, bilateral e simétrica, sem associação com inflamação ocular prévia. Distrofias corneanas são classificados de acordo com a camada corneana envolvida em distrofia superficial, estromal e posterior. A incidência de cada distrofia varia de acordo com a região geográfica estudada. OBJETIVO: Avaliar a prevalência de distrofias corneanas estromal em botões corneanos de espécimes obtidos por ceratoplastia penetrante (CP), oriundos do arquivo de um laboratório de patologia ocular e correlacionar o diagnóstico com a idade e o sexo dos pacientes. MÉTODOS: Os botões corneanos oriundos de ceratoplastia penetrante recebidos entre janeiro de 1996 e maio de 2009 foram selecionados dos arquivos do Henry C. Witelson Ocular Pathology and Registry Laboratory, em Montreal, Canadá. Os casos com diagnóstico histopatológico de distrofias corneanas estromal foram corados com colorações especiais ("Peroxid acid Schiff", tricrômico de Masson, vermelho Congo analisadas sob luz polarizada, e "alcian blue") para a classificação e foram correlacionados com dados epidemiológicos (idade na época da ceratoplastia penetrante e sexo) dos pacientes. RESULTADOS: 1.300 casos de botões corneanos com diagnóstico clínico de distrofia corneana foram recuperados. Distrofia corneana estromal foi encontrada em 40 (3,1%) dos casos. Distrofia corneana lattice foi a mais prevalente com 26 casos (65%). Dezenove eram do sexo feminino (73,07%) e CP foi realizada em média com 59,3 anos de idade. Distrofia corneana combinada foi encontrada em 8 (20%) casos, 5 (62,5%) eram do sexo feminino e a idade média da CP foi de 54,8 anos. Distrofia corneana granular foi encontrada em 5 (12,5%) casos, e 2 (40%) deles eram do sexo feminino. A ceratoplastia penetrante foi realizada na média de idade de 39,5 anos, em casos de distrofia corneana granular. A distrofia corneana macular esteve presente em apenas um caso (2,5%), 36 anos de idade do sexo feminino. CONCLUSÃO: A abordagem histopatológica e avaliação sistemáticas, incluindo colorações especiais em todas as distrofias corneanas é essencial para estabelecer o correto diagnóstico.
Descritores: Distrofias hereditárias da córnea; Distrofias hereditárias da córnea; Ceratoplastia penetrante; Substância própria
ORIGINAL ARTICLE ARTIGO ORIGINAL
Corneal stromal dystrophies: a clinical pathologic study
Distrofia corneana estromal: um estudo clínicopatológico
Elvira Barbosa AbreuI; Gustavo Amorin NovaesII; Bruno Franco FernandesII; Patricia Rusa Pereira OdashiroII; Alexandre Nakao OdashiroII; Isabella de Oliveira Lima ParizottoIII; Miguel Noel Burnier Jr. II
IPhysician, Instituto Penido Burnier, Campinas (SP) - Brasil
IIPhysician, Henry C. Witelson Ocular Pathology Laboratory - McGill University, Montreal-QC, Canada
IIIMedical Student. Henry C. Witelson Ocular Pathology Laboratory - McGill University, Montreal-QC, Canada
ABSTRACT
INTRODUCTION: Corneal dystrophy is defined as bilateral and symmetric primary corneal disease, without previous associated ocular inflammation. Corneal dystrophies are classified according to the involved corneal layer in superficial, stromal, and posterior dystrophy. Incidence of each dystrophy varies according to the geographic region studied.
PURPOSE: To evaluate the prevalence of stromal corneal dystrophies among corneal buttons specimens obtained by penetrating keratoplasty (PK) in an ocular pathology laboratory and to correlate the diagnosis with patient age and gender.
METHODS: Corneal button cases of penetrating keratoplasty from January-1996 to May-2009 were retrieved from the archives of The Henry C. Witelson Ophthalmic Pathology Laboratory and Registry, Montreal, Canada. The cases with histopathological diagnosis of stromal corneal dystrophies were stained with special stains (Peroxid acid Schiff, Masson trichrome, Congo red analyzed under polarized light, and alcian blue) for classification and correlated with epidemiological information (age at time of PK and gender) from patients' file.
RESULTS: 1,300 corneal buttons cases with clinical diagnose of corneal dystrophy were retrieved. Stromal corneal dystrophy was found in 40 (3.1%) cases. Lattice corneal dystrophy was the most prevalent with 26 cases (65%). Nineteen were female (73.07%) and the PK was performed at average age of 59.3 years old. Combined corneal dystrophy was found in 8 (20%) cases, 5 (62.5%) of them were female and the average age of the penetrating keratoplasty was 54.8 years old. Granular corneal dystrophy was represented by 5 (12.5%) cases, and 2 (40%) of them were female. Penetrating keratoplasty was performed at average age of 39.5 years old in granular corneal dystrophy cases. Macular corneal dystrophy was present in only 1 (2.5%) case, in a 36 years old female.
CONCLUSION: Systematic histopathological approach and evaluation, including special stains in all stromal corneal dystrophies is critical to establish the correct diagnosis.
Keywords: Corneal dystrophies, hereditary/pathology; Corneal dystrophies, hereditary/epidemiology; Keratoplasty, penetrating; Corneal stroma
RESUMO
INTRODUÇÃO: A distrofia corneana é definida como doença primária da córnea, bilateral e simétrica, sem associação com inflamação ocular prévia. Distrofias corneanas são classificados de acordo com a camada corneana envolvida em distrofia superficial, estromal e posterior. A incidência de cada distrofia varia de acordo com a região geográfica estudada.
OBJETIVO: Avaliar a prevalência de distrofias corneanas estromal em botões corneanos de espécimes obtidos por ceratoplastia penetrante (CP), oriundos do arquivo de um laboratório de patologia ocular e correlacionar o diagnóstico com a idade e o sexo dos pacientes.
MÉTODOS: Os botões corneanos oriundos de ceratoplastia penetrante recebidos entre janeiro de 1996 e maio de 2009 foram selecionados dos arquivos do Henry C. Witelson Ocular Pathology and Registry Laboratory, em Montreal, Canadá. Os casos com diagnóstico histopatológico de distrofias corneanas estromal foram corados com colorações especiais ("Peroxid acid Schiff", tricrômico de Masson, vermelho Congo analisadas sob luz polarizada, e "alcian blue") para a classificação e foram correlacionados com dados epidemiológicos (idade na época da ceratoplastia penetrante e sexo) dos pacientes.
RESULTADOS: 1.300 casos de botões corneanos com diagnóstico clínico de distrofia corneana foram recuperados. Distrofia corneana estromal foi encontrada em 40 (3,1%) dos casos. Distrofia corneana lattice foi a mais prevalente com 26 casos (65%). Dezenove eram do sexo feminino (73,07%) e CP foi realizada em média com 59,3 anos de idade. Distrofia corneana combinada foi encontrada em 8 (20%) casos, 5 (62,5%) eram do sexo feminino e a idade média da CP foi de 54,8 anos. Distrofia corneana granular foi encontrada em 5 (12,5%) casos, e 2 (40%) deles eram do sexo feminino. A ceratoplastia penetrante foi realizada na média de idade de 39,5 anos, em casos de distrofia corneana granular. A distrofia corneana macular esteve presente em apenas um caso (2,5%), 36 anos de idade do sexo feminino.
CONCLUSÃO: A abordagem histopatológica e avaliação sistemáticas, incluindo colorações especiais em todas as distrofias corneanas é essencial para estabelecer o correto diagnóstico.
Descritores: Distrofias hereditárias da córnea/patologia; Distrofias hereditárias da córnea/epidemiologia; Ceratoplastia penetrante; Substância própria
INTRODUCTION
Corneal dystrophy (CD) is defined as bilateral and symmetric primary corneal disease, without previous associated ocular inflammation. Most cases of CD have an autosomal dominant inheritance pattern, starting at the first decades of life, with a stable or slowly progressive course(1,2).
Corneal dystrophies (CDs) are classified according to the involved corneal layer. Superficial CDs include Messman's, gelatinous, Lisch's epithelial, mucinous subepithelial, Reis-Bucklers, Thiel-Benke dystrophy and recurrent epithelial erosion. Stromal CDs include macular, granular, reticular (lattice), Schnyder's, "fleck", congenital stromal, and posterior amorphous dystrophy. Posterior CDs include Fuchs, posterior polymorphous, congenital endothelial, and endothelial X linked dystrophy(3,4).
Different genetic mutations have been related with CDs development. Most frequently involved genes are: TGFBI, CHST6, KRT3, KRT 12, PIP 5k3, SLC 4AIII, TATICSt2, and UBIADI(5,6).
Stromal CDs are related to mutated keratoepithelin formation, except the macular type in witch mutation is located at carbohydrate sulfotransferase 6 gene (CHST6). This protein has the diffusing capacity through Bowman's layer and stroma, where it aggregates to other substances, forming deposits(3,5).
Incidence of each dystrophy varies according to the geographic region studied(7-12). According to the literature, CDs prevalence in corneal buttons from penetrating keratoplasty (PK) varies from 1.3% to 4.0%(13-16). Others studies have shown a higher prevalence of CDs, such as 12.6% in Japan and 23.2% in France(10,17).
Diagnosis is usually suspected during biomicroscopy examination, and confirmed with histopathology exam, after PK. In special cases, genetic studies can be required for diagnoses.
The diagnostic histopathological exam of stromal CDs include routine hematoxilin-eosin and special histochemical stains such as peroxid acid Schiff, Masson trichrome, Congo red and alcian blue(2).
This study was performed to evaluate the prevalence of stromal CDs among corneal buttons specimens obtained by PK in an ocular pathology laboratory and to correlate the diagnosis with patient age and gender.
METHODS
All corneal button cases of PK from January-1996 to May-2009 were retrieved from the archives of The Henry C. Witelson Ophthalmic Pathology Laboratory and Registry, Montreal, Canada.
The cases with histopathological diagnosis of stromal CDs were stained with special stains (Peroxid acid Schiff, Masson trichrome, Congo red analyzed under polarized light, and alcian blue) for classification according to a standard scheme (Table 1). The cases were correlated with epidemiological information available (age at time of PK and gender) from patients' file.

RESULTS
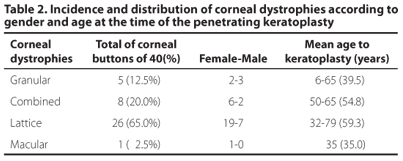
A total of 1,300 corneal buttons cases with clinical diagnose of corneal dystrophy were retrieved. Stromal CD was found in 40 (3.1%) cases. Lattice CD was the most prevalent with 26 cases (65%). Nineteen were female (73.07%) and the PK was performed at average age of 59.3 years old.
Combined CD was found in 8 (20%) cases, 5 (62.5%) of them were female and the average age of the PK was 54.8 years old.
Granular CD was represented by 5 (12.5%) cases, and 2 (40%) of them were female. PK was performed at average age of 39.5 years old in Granular CD cases.
Macular CD was present in only one (2.5%) case, in a 36 years old female.
Results are summarized on table 2.
DISCUSSION
Corneal dystrophies are a group of heterogenic diseases, spontaneous, bilateral and progressive, without inflammatory signs, genetically transmitted and, usually without systemic associations. Most of them have an autosomal dominant pattern of inheritance, starting at the first decades of life, with a stable or slowly progressive course(2).
Clinically, they are classified according to the corneal layer involved, and can be divided in 3 groups: Anterior or superficial dystrophies, where the epithelium, the Bowman's layer and the superficial stroma are primarily involved. Stromal corneal dystrophies, in which the stroma is affected (subject of this study). Posterior dystrophies involve the Descemet's membrane and the endothelium(4).
We are going to a quick review the most clinical relevant stromal dystrophies.
Granular dystrophy
Granular dystrophy, also known as Groenouw type I, is an autosomal dominant disease related to the transforming growth factor beta (TGFB1) gene and 5q31 gene locus. Biomicroscopically, white, well-defined granules with a crushed bread crumbs appearance can be seen. The opacities do not extend all the way to the limbus and as the diseases progresses these granules may extend into the deeper stroma down to Descemet membrane. Corneal deposits can occur as early as 2 years of age, but rarely visual acuity is impaired before older age. Early symptoms include glare and photophobia. As the condition evolves and the granules become more confluent, decrease in vision and recurrent erosions with pain may occur. On histopathology, it appears as multiple stromal deposits that may extend from deep epithelium to Descemet membrane. The hyaline opacities stain positively with Masson trichrome. Transmission microscopy can show rod shaped bodies similar to the ones found at Reis- Bucklers dystrophy. That is why some authors speculate that the later might be a superficial variant of granular dystrophy(3,4).
Combined granular-lattice dystrophy
Also know as Avellino's corneal dystrophy, combined dystrophy exhibits features of both granular and lattice dystrophy. It is also inherited in an autosomal dominant fashion and related to the TGFB1 gene, locus 5q31. It was primarily termed as Avellino dystrophy because histopathological analysis of families affected had ancestors in the region of Avellino, Italy (18,19). Later reports revealed many other cases of patients from other nationalities, without history of Italian ancestors. Biomicroscopically, granular deposits are more superficial and as the disease progresses, a snowflake appearance deeper in the stroma can be noted. The linear refractile deposits tend to be deeper than the granular deposits, but with progression these lines coalesce with the round opacities. First signs of the disease can be seen as early as 3 years of age in homozygotic cases, but most commonly during puberty or early adulthood. Visual acuity deteriorates as the central visual axis becomes affected and corneal erosions can result in episodes of pain. Even though, it usually progresses slowly, homozygotes can have a more rapid course comparing with heterozygotes patients. On histopathological exam, corneal opacities extend from the basal epithelium to the deep stroma and individual opacities stain positive with either Masson trichrome or Congo red(3,4).
Lattice dystrophy
Lattice dystrophy can be classified in 4 variants. Type I, the classic form also known as Biber-Haab-Dimmer, has autosomal dominant inheritance involving the gene TGFB1 in the locus 5q31. It manifests in childhood and does not present systemic associations. Type II, also known as Meretoja's syndrome, is associated with systemic amiloidosis. This type has also autosomal dominant inheritance and mutation is located in the gelsolin gene, at 9q chromosome. In types III and IV inheritance patterns vary. In type III, for example, inheritance is recessive. They also vary in clinical manifestations, initiating earlier or later in life, presenting reticular thin or greater deposits. In classic form here studied, biomicroscopy reveals initially, discrete sub-epithelial opacities, ovoid or round, white points in anterior stroma and small refractive lines that appear, usually, at the first decade of life. As the condition progresses, lesions change their aspect, appearing as small nodules, dots or thick and branched lines progressing to the deeper stroma and to the epithelium. At this point, stroma between the lesions is clear, but with evolution, opacities tend to coalesce and anterior and medium stroma may be diffusely opaque. On histopathological examination, amyloid deposits stain positive with Congo red. Green birefringence is visible with a polarizing filter and red-green dichroism when a green filter is added with this stain. Metachromasia is noted with crystal violet and fluorescence is noted with use of thioflavin T staining(3,4).
Macular
Macular dystrophy, also known as Groenouw corneal dystrophy type II, unlike most of the corneal dystrophies, is inherited in an autosomal recessive fashion, involving the gene CHST6 in the 16q22 locus. Early in the disease, biomicroscopy shows a diffuse stromal haze extending to the limbus. As the disease progresses, superficial, central, elevated, and irregular whitish opacities (macules) develop. Posterior stroma can also be involved. There are no clear areas between the opacities and the cornea is usually thinner than normal. Later in the course, the endothelium can be involved and decrease in its functions can thicken the cornea. Corneal findings appear early in life and vision can be severely impaired between 10 and 30 years of age, even though it is a slowly progressive disease. Histopathology shows Glycosaminoglycans (GAGs) accumulation intra and extracellularly in corneal stroma, Descemet's membrane and endothelium. Guttae are often present at the Descemet membrane. GAGs stain positively with Hale colloidal iron or alcian blue(3,4).
Studies published in different continents showed that the dystrophies incidence can vary according to the geographic area studied. In Saudi Arabia and India, the most common corneal stromal dystrophy is macular, with recessive inheritance, that might be explained by high taxes of consanguineous weddings in that population(7,8). In 1995, found a discrete prevalence of lattice dystrophy in relation to granular(17). In a paper published in 1987, Lang et al. , compared penetrating keratoplasty indications in the United States and in Europe, observing that, in the United States, the most frequent stromal dystrophy among the studied specimens was granular dystrophy(9).
A very extensive study, made at Unifesp, in1990, found among 1,000 corneas examined, 15 granular dystrophies, 11 macular dystrophies and 11 lattice dystrophies(20).
As personal communication, in 638 patients with corneal transplant and histopathological examination in Campinas, São Paulo, Brazil, we found 15 corneal stromal dystrophies (2.35%). Nine were granular dystrophy (60%), 4 were lattice dystrophy (26.7%) and 2 were macular dystrophies (13.3%). Both cases of macular dystrophy were present in brothers of a numerous family, with consanguinity history, which explains the occurrence of this dystrophy in the region. However, the histopathological exam of all cases was performed by different pathologists from different institutions and without a standard method. Clinical characteristics, age of presentation and progression are usually similar among members of the same family, but genetic defect expression can vary, resulting in differences in disease's natural history(21).
In this study, there are 2 children, a 6-year-old and a 10-year-old, submitted to penetrating keratoplasty very early. It is probable that these patients are homozygous for the tgfbi mutation, presenting more severe dystrophy forms or that genetic penetration in these cases was complete(22,23).
In granular dystrophy, the average age of patients submitted to penetrating keratoplasty is 48.4 years old(9). In this present study, there was no clear predilection for any gender, with slightly more common incidence among males. This observation is in accordance with other authors(24). Some authors described the coexistence of amyloid deposits in cases previously thought to be granular dystrophy(25). This observation can lead us to think that probably some of previous cases of granular dystrophy reported were, in fact combined dystrophies not diagnosed yet.
In lattice dystrophy, the average age at the time that penetrating keratoplasty is around 40 years old and some authors show prevalence in male patients(26-28). However, in one study of 67 patients with lattice dystrophy, no difference in gender was found(26). The rate of lattice dystrophy was higher in this study compared to the others described in the literature. This could be explained by the worldwide diversity of population settled in Montreal.
At the present study, we found that the observation of amyloid deposits in corneal buttons studied was frequent, reaching 85% (65% lattice and 20% combined dystrophies). We also found that 17.5% of clinical diagnostic hypothesis did not correspond to histopathological findings. All 7 examined corneas presented positive stain for amyloid, thus when stained by Congo red and studied under polarized light deposits presented dichroism, showing apple-green coloration. In 4 of them, clinical diagnosis was granular dystrophy and in 3, the hypothesis was lattice dystrophy. Probably in these 3 last cases the hypothesis was formulated in late stages of the disease, when corneal haze is already very intense. So far, it has not been described cases in which lattice lesions appear alone or previously than the granular ones in combined dystrophy. In the other four cases previously diagnosed as granular dystrophy, probably happened exactly what other authors already have described: clinically, patients presenting only granular deposits, but histopathological exam revealing associated amyloid deposits(29).
Because we only had one case of macular dystrophy, any conclusion about epidemiological data could be misleading. In 1987, 16 cases of macular dystrophy were reported and it showed to be related to younger age (mean of 43 yo) at the time of penetrating keratoplasty among the stromal dystrophies. Female gender was predominant(9).
In this study, we did not find any case of central crystalline dystrophy (Schnider), gelatinous drop, congenital stromal dystrophy, polymorphous stromal dystrophy, marginal crystalline dystrophy (Bietti) or posterior amorphous dystrophies.
To our knowledge, this is the largest stromal dystrophy series studied in a single ocular pathology laboratory, with a standardized method of evaluation for the disease, previously diagnosed by anterior segment specialists of the same institution.
CONCLUSION
Despite rare among the different corneal diseases, dystrophies involving amyloid deposition are the most frequent. Systematic histopathological approach and evaluation, including special stains in all stromal corneal dystrophies is critical to establish the correct diagnosis. All corneal buttons with clinical diagnosis of stromal dystrophy should be sent to histopathological analysis to confirm the diagnosis.
REFERENCES
1. Malbran ES. Corneal dystrophies: a clinical, pathological, and surgical approach. 28 Edward Jackson Memorial Lecture. Am J Ophthalmol. 1972;74(5):771-809.
2. Waring GO, 3rd, M.M. Rodrigues, Laibson PR. Corneal dystrophies. I. Dystrophies of the epithelium, Bowman's layer and stroma. Surv Ophthalmol. 1978;23(2):71-122.
3. Weiss JS, Moller HU, Lisch W, Kinoshita S, Aldave AJ, Belin MW, et al. The IC3D classification of the corneal dystrophies. Cornea. 2008;27 Suppl 2:S1-83. Comment in: Cornea. 2010;29(12):1469.
4. Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7.
5. Bron AJ. Genetics of the corneal dystrophies: what we have learned in the past twenty-five years. Cornea. 2000;19(5):699-711.
6. Poulak V, Colby K. Genetics of anterior and stromal corneal dystrophies. Semin Ophthalmol. 2008;23(1): 9-17.
7. Pandrowala H, Bansal A, Vemuganti GK, Rao GN. Frequency, distribution, and outcome of keratoplasty for corneal dystrophies at a tertiary eye care center in South India. Cornea. 2004;23(6):541-6.
8. al Faran MF, Tabbara KF. Corneal dystrophies among patients undergoing keratoplasty in Saudi Arabia. Cornea. 1991;10(1):13-6.
9. Lang GK, Naumann GO. The frequency of corneal dystrophies requiring keratoplasty in Europe and the U.S.A. Cornea. 1987;6(3):209-11.
10. Leger F, Ndiaye PA, Williamson W, Lagoutte F, Riss I. [Indications of penetrating keratoplasty from a histopathological study of 1129 corneal buttons (from 1982 to 1991)]. J Fr Ophtalmol. 1995;18(5):331-7. French.
11. Rozzo C, Fossarello M, Galleri G, Sole G, Serru A, Orzalesi N, et al. A common beta ig-h3 gene mutation (delta f540) in a large cohort of Sardinian Reis Bucklers corneal dystrophy patients. Mutations in brief no. 180. Online. Hum Mutat. 1998;12(3): 215-6.
12. Shah SS, Al-Rajhi A, Brandt JD, Mannis MJ, Roos B, Sheffield VC, et al. Mutation in the SLC4A11 gene associated with autosomal recessive congenital hereditary endothelial dystrophy in a large Saudi family. Ophthalmic Genet. 2008;29(1):41-5.
13. Al-Yousuf N, Mavrikakis I, Mavrikakis E, Daya SM. Penetrating keratoplasty: indications over a 10 year period. Br J Ophthalmol. 2004;88(8):998-1001.
14. Maeno A, Naor J, Lee HM, Hunter W, Rootman DS. Three decades of corneal transplantation: indications and patient characteristics. Cornea. 2000;19(1):7-11.
15. Cosar CB, Sridhar MS, Cohen EJ, Held EL, Alvim P de T, Rapuanao CJ, et al. Indications for penetrating keratoplasty and associated procedures, 1996-2000. Cornea. 2002; 21(2):148-51.
16. Santos LN, Fernandes BF, de Moura LR, Cheema DP, Maloney S, Logan P, et al. Histopathologic study of corneal stromal dystrophies: a 10-year experience. Cornea. 2007; 26(9):1027-31.
17. Santo RM, Yamaguchi T, Kanai A, Okisaka S, Nakajima A. Clinical and histopathologic features of corneal dystrophies in Japan. Ophthalmology. 1995;102(4):557-67.
18. Holland EJ, Daya SM, Stone EM, Folberg R, Dobler AA, Cameron JD, et al. Avellino corneal dystrophy. Clinical manifestations and natural history. Ophthalmology. 1992; 99(10):1564-8.
19. Folberg R, Stone EM, Sheffield VC, Mathers WD. The relationship between granular, lattice type 1, and Avellino corneal dystrophies. A histopathologic study. Arch Ophthalmol. 1994;112(8):1080-5.
20. Cunha MC. Contribuição ao estudo das distrofias corneanas [tese]. São Paulo: Universidade Federal de São Paulo; 1990.
21. Solari HP. Estudo do gene TGFBI em pacientes com distrofia de córnea. [tese]. São Paulo: Universidade Federal de São Paulo; 2005.
22. Konishi M, Yamada M, Nakamura Y, Mashima Y. Varied appearance of cornea of patients with corneal dystrophy associated with R124H mutation in the BIGH3 gene. Cornea. 1999;18(4):424-9.
23. Akimune C, Watanabe H, Maeda N, Okada M, Yamamoto S, Kiritoshi A, et al. Corneal guttata associated with the corneal dystrophy resulting from a betaig-h3 R124H mutation. Br J Ophthalmol. 2000;84(1):67-71.
24. Moller HU, Ehlers N. Early treatment of granular dystrophy (Groenouw type I). Acta Ophthalmol (Copenh). 1985;63(5):597-600.
25. Folberg R, Alfonso E, Croxatto JO, Driezen NG, Panjwani N, Laibson PR, et al. Clinically atypical granular corneal dystrophy with pathologic features of lattice-like amyloid deposits. A study of these families. Ophthalmology. 1988;95(1):46-51.
26. Meisler DM, Fine M. Recurrence of the clinical signs of lattice corneal dystrophy (type I) in corneal transplants. Am J Ophthalmol. 1984;97(2):210-4.
27. Hida T, Tsubota K, Kigasawa K, Murata H, Ogata T, Akiya S. Clinical features of a newly recognized type of lattice corneal dystrophy. Am J Ophthalmol. 1987;104(3):241-8.
28. Lang GK, Wilk CM, Naumann GO. [Changes in the indications status for keratoplasty (Erlangen, 1964-1986)]. Fortschr Ophthalmol. 1988;85(3):255-8. German.
29. Konishi M, Mashima Y, Nakamura Y, Yamada M, Sugiura H. Granular-lattice (Avellino) corneal dystrophy in Japanese patients. Cornea. 1997;16(6):635-8.
 Correspondence address:
Correspondence address:
Elvira Barbosa Abreu. 3775
University Street Room 216
H3A 2B4 Montreal - QC, Canada
E-mail: [email protected]
Submitted for publication: March 14, 2012
Accepted for publication: September 20, 2012
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: E.B. Abreu, None; G.A. Novaes, None; B.F. Fernandes, None; P.R.P. Odashiro, None; A.N. Odashiro, None; I.O.L.Parizotto, None; M.N.Burnier Jr, None.
Work should be assigned to: Henry C. Witelson Ocular Pathology Laboratory - McGill University, Montreal-QC, Canada.
© 2024 - All rights reserved - Conselho Brasileiro de Oftalmologia
![]()
 English PDF
English PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Scielo
Scielo
 Pocket
Pocket
