Arq. Bras. Oftalmol. 2022;85 (6 )
:584-589
| DOI: 10.5935/0004-2749.20220089
Abstract
Objetivo: Determinar os efeitos da deficiência de vitamina D nos microvasos da retina usando angiotomografia de coerência óptica.
Métodos: Este estudo foi planejado para ser do tipo caso-controle observacional. Foram avaliados 98 olhos de pacientes com deficiência de vitamina D e 96 olhos de participantes saudáveis com nível sérico de vitamina D superior a 30 ng/mL. Foram adquiridas imagens de varredura centralizadas na mácula, com um tamanho de 6,00 × 6,00 mm. Mediram-se a densidade dos vasos nos plexos capilares superficial e profundo da retina, a área da zona avascular foveal e a área do fluxo coriocapilar.
Resultados: Os grupos mostraram-se semelhantes em relação à melhor acuidade visual corrigida, ao gênero, ao comprimento axial, ao erro refrativo, à idade e à pressão intraocular ajustada. O nível médio de vitamina D foi significativamente menor no grupo de estudo (p=0,021). As densidades total, parafoveal e perifoveal do plexo capilar profundo foram significativamente maiores no grupo de estudo que no grupo controle (respectivamente, p=0,012, p=0,014 e p=0,023). As áreas da zona avascular foveal e do fluxo coriocapilar foram semelhantes nos dois grupos (respectivamente, p=0,37 e p=0,27). Além disso, houve uma forte correlação negativa do nível sérico de vitamina D com as densidades vasculares medidas em toda a imagem e nas regiões parafoveais e perifoveais do plexo capilar profundo no grupo de estudo (respectivamente, ρ de Spearman = -0,71, p=0,043; ρ de Spearman = -0,79, p=0,011; e ρ de Spearman = -0,74, p=0,032).
Conclusão: Pode ocorrer um aumento na densidade vascular da retina devido a alterações estruturais dos vasos causadas pela deficiência de vitamina D. O aumento da densidade vascular, especialmente no plexo capilar profundo, pode ser usado para o diagnóstico precoce da vasculopatia associada à deficiência de vitamina D.
Keywords: Deficiência de vitamina D; Vasos retinianos/fisiopatologia; Doenças vasculares/ prevenção & controle; Tomografia de coerência óptica
Arq. Bras. Oftalmol. 2021;84 (4 )
:367-373
| DOI: 10.5935/0004-2749.20210053
Abstract
OBJETIVO: A doença de Stargardt é a forma mais comum de distrofia macular de início juvenil. É bilateral e simétrica em aparência, afeta a mácula e sua característica principal é a diminuição da visão central que geralmente inicia-se na primeira ou segunda década de vida. O objetivo do estudo é descrever o perfil clínico dos pacientes avaliados no Complexo Hospital de Clínicas da Universidade Federal do Paraná, bem como descrever os achados eletrorretinográficos destes pacientes com o eletrorretinograma de campo total.
MÉTODOS: Foi realizado um estudo observacional retrospectivo, baseado na análise de prontuários e eletrorretinograma de 27 pacientes com Doença de Stargardt e Fundus Flavimaculatus, atendidos em consulta oftalmológica no ambulatório de Eletrofisiologia Ocular e Neuro-Oftalmologia do Complexo Hospital de Clínicas da Universidade Federal do Paraná, entre 1997 e 2014. Os pacientes incluídos no estudo apresentavam quadro clínico, fundoscopia e/ou achados eletrorretinográficos compatíveis com a doença.
RESULTADOS: A acuidade visual no melhor olho variou de 0 a 1,6 logMAR (20/20 a 20/800), com média de 0,89 ± 0,42 logMAR. A idade de aparecimento dos sintomas variou desde o nascimento a 36 anos (19,2 ± 9,2), sendo a maioria nas 1ª e 2ª década de vida. Em relação ao tempo entre o início dos sintomas e o diagnóstico, a média foi de 7,3 anos. Na fundoscopia, todos os pacientes apresentaram alguma alteração. Na análise do eletrorretinograma, a maioria dos pacientes demonstrou resultados que diferem da amostra de pacientes controles, ou seja, amplitudes reduzidas e tempos de culminação aumentados nas fases fotópicas e escotópicas.
CONCLUSÕES: A acuidade visual e idade de início de aparecimento dos sintomas encontrados neste estudo são compatíveis com a evolução desta distrofia. Achados fundoscópicos típicos da doença de Stargardt e eletrorretinograma alterados foram mais frequentes em decorrência do atraso no diagnóstico. Novos estudos prospectivos são necessários para avaliar estes pacientes, fundamentando-se em novas tecnologias.
Keywords: Eletrorretinografia; Doenças retinianas; Epitélio pigmentado da retina; Degeneração macular; Lipofuscina
Arq. Bras. Oftalmol. 2025;88 (3 )
:1-8
| DOI: 10.5935/0004-2749.2023-0115
Abstract
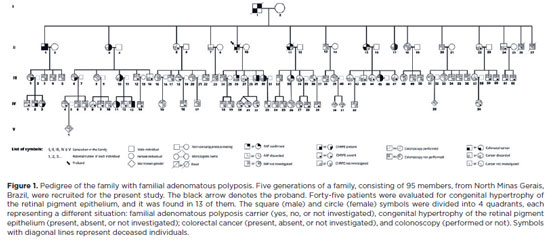
PURPOSE: To evaluate the presence of congenital hypertrophy of the retinal pigment epithelium in a large family affected by familial adenomatous polyposis and identify the causative mutation in the adenomatous polyposis coli gene. Thus, we aimed to determine the significance of congenital hypertrophy of the retinal pigment epithelium as a phenotypic marker of the disease.
METHODS: A family consisting of 95 individuals was evaluated. Among these, 45 individuals were randomly selected by convenience sampling method to undergo ophthalmological evaluation. A funduscopic exam, including slit lamp and indirect ophthalmoscopy, were performed in the selected patients. In those with retinal lesions, a retinography was obtained. The adenomatous polyposis coli gene was sequenced in one affected family member to identify the pathogenic mutation. Once the variant was identified, six undiagnosed family members were tested for the mutation via capillary electrophoresis sequencing.
RESULTS: Congenital hypertrophy of the retinal pigment epithelium was observed in 13 (28.9%) of the 45 individuals evaluated. Of these, nine patients were confirmed to have familial adenomatous polyposis (via colonoscopy or molecular testing). However, four patients had not been investigated. Of the 32 (71.1%) family members without the lesion, 14 did not have familial adenomatous polyposis and 18 were yet to be evaluated. The lesions were bilaterally present and exhibited a peculiar fish-tail shape in all the evaluated individuals. Adenomatous polyposis coli gene sequencing revealed a pathogenic variant c.4031del. (Ser1344*), in heterozygosity (49.27%), in exon 16.
CONCLUSIONS: The study findings confirmed the significance of congenital hypertrophy of the retinal pigment epithelium as a phenotypic marker for familial adenomatous polyposis. Furthermore, it is an effective first-line screening method for at risk family members of such patients. The novel mutation identified in our study participants, which is yet to be described in the literature, causes an aggressive form of the disease.
Keywords: Retinal diseases/congenital; Retinal pigment epithelium; Hypertrophy/congenital; Adenomatous polyposis coli / genetics; Phenotype; Optical coherence tomography
Arq. Bras. Oftalmol. 2024;87 (4 )
:1-8
| DOI: 10.5935/0004-2749.2021-0415
Abstract
Objetivo: Fenótipos Stargardt-like já foram associados a variantes patogênicas no gene ABCA4. O propósito desse estudo é descrever quatro pacientes com achados retinianos semelhantes a doença de Stargardt com resultados moleculares diferentes do esperado.
Métodos: Esse relato fez a revisão de prontuários médicos de quatro pacientes com distrofia macular e achados clínicos sugestivos de doença de Stargardt. Foram realizados avaliação oftalmológica, exames de imagens e testes usando next generation sequencing para avaliar variantes patogênicas associadas aos fenótipos dos pacientes.
Resultados: Os pacientes apresentavam atrofia macular e alterações pigmentares sugerindo achados clínicos de doença de Stargardt. Dois pacientes foram associados a genes com herança autossômica dominante (RIMS1 e CRX) e dois pacientes foram associados a genes com herança autossômica recessiva (CRB1 e RDH12) com variantes preditoras de serem patogênicas.
Conclusão: Distrofias maculares podem ter similaridades fenotípicas com fenótipo de Stargardt-like associados a outros genes além dos classicamente já descritos.
Keywords: Doença de Stargardt; Estudos de associação genética; Fenótipo; Padrões de herança; Sequenciamento de nucleotídeos em larga escala; Degeneração macular; Distrofias retinianas; Doenças genéticas
Arq. Bras. Oftalmol. 2025;88 (3 )
:1-8
| DOI: 10.5935/0004-2749.2024-0104
Abstract
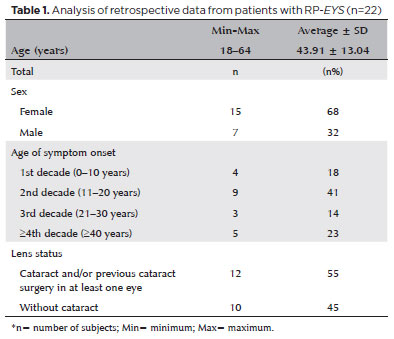
PURPOSE: This study aimed to characterize retinitis pigmentosa associated with the eyes shut homolog gene, which causes hereditary retinal degeneration.
METHODS: The anatomical and functional findings of retinitis pigmentosa in patients with variants of the eyes shut homolog gene were characterized and compared using multimodal imaging and genetic analysis of the variants. Clinical data such as visual acuity, lens status, and refraction were obtained from medical records. Patients underwent an ophthalmic examination, including static visual field, microperimetry, optical coherence tomography, fundus autofluorescence, and fundus photography.
RESULTS: Twenty-two patients were included in the study. Several anatomical and functional characteristics of retinitis pigmentosa-eyes shut homolog were identified, including the presence of cataracts, cystoid macular edema, epiretinal membrane, and a tubular visual field. Genetic results revealed 26 distinct variants in the cohort, with 7 novel variants not previously documented or reported in the scientific literature or databases.
CONCLUSION: The findings demonstrate that eyes shut homolog-retinitis pigmentosa manifests in specific patterns, starting in adolescence with mild progression and advancing with age. The integration of multimodal imaging and genetic analysis has provided a detailed understanding of the anatomical and functional features of retinitis pigmentosa-eyes shut homolog. Seven novel variants of the eyes shut homolog gene have been identified. These findings enhance the understanding of eyes shut homolog-related retinitis pigmentosa characteristics of by detailing the spectrum of mutations in this gene within the Brazilian population.
Keywords: Retinal diseases/diagnostic imaging; Retinitis pigmentosa/genetics; Retinal degeneration; Eye proteins/genetics; Eye diseases, hereditary/genetics; Genes, recessive; Phenotype; Multimodal imaging; Tomography, optical coherence/methods; Fluorescein angiogr
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
14-fig01.jpg)
10-tab01tb.jpg)
05-tab01tb.jpg)
04-fig01.jpg)
04-fig01tb.jpg)
02-fig01.jpg)
14-fig01.jpg)
03-fig01.jpg)
03-fig01.jpg)
12-fig01.jpg)
15-fig01.jpg)