Arq. Bras. Oftalmol. 2020;83 (6 )
:473-477
| DOI: 10.5935/0004-2749.20200088
Abstract
Objetivos: Descrever os achados na angiografia por tomografia de coerência óptica associada à síndrome de Alport.
Métodos: Estudo descritivo de um serviço de referência em Oftalmologia (Hospital Evangélico de Vila Velha, Brasil). Os pacientes diagnosticados com síndrome de Alport, foram incluídos.
Resultados: O grupo de estudo foi composto por quatro pacientes (um feminino e três homens) com diagnóstico de síndrome de Alport. A acuidade visual no pior olho estava entre 20/40 a 20/60. Todos os pacientes do sexo masculino apresentaram lenticone anterior à biomiscroscopia. Os achados da retina observados incluíram pontos e manchas e alterações pigmentares na mácula. Na angiotomografia de coerência óptica, as camadas internas da retina de todos os pacientes apresentaram afinamento (especialmente na região temporal da mácula) e aumento da zona avascular foveal. Uma coroide espessa foi observada em ambos os olhos dos dois pacientes mais jovens.
Conclusões: Em pacientes com síndrome de Alport, as camadas internas da retina sofrem alterações devido à mutação do colágeno tipo IV. A angiotomografia de coerência óptica permite visualizar esses achados, tornando-o uma ferramenta útil na detecção de achados iniciais da retina associados à síndrome de Alport.
Keywords: Retina; Tomografia de coerência óptica; Angiofluoresceínografia/métodos; Nefrite hereditária
Arq. Bras. Oftalmol. 2022;85 (1 )
:13-18
| DOI: 10.5935/0004-2749.20220003
Abstract
Objetivo: Fornecer informações sobre a ocorrência e a eficácia do aconselhamento sobre o uso de tabaco por oftalmologistas a pacientes com doenças oculares associadas à tireoide.
Métodos: Analisamos os prontuários médicos eletrônicos de uma coorte digital de pacientes atendidos por oftalmologistas no Sistema de Saúde da Universidade da Pensilvânia entre o início de 2012 e o final de 2017 com os códigos da Classificação Internacional de Doenças (CID) para a doença de Graves, exoftalmia tireotóxica ou doença ocular associada à tireoide. Os históricos de uso de tabaco foram registrados na primeira e na última visita ao consultório de Oftalmologia, ou na visita mais próxima no tempo. A quantidade de maços/dia (mpd) e todas as anotações feitas nas visitas ao consultório de Oftalmologia foram analisadas para aconselhamento sobre o uso de tabaco.
Resultados: Um total de 435 indivíduos preencheram os critérios de inclusão, dos quais 72 (16,6%) estavam fumando ativamente no momento do primeiro encontro. Apenas 57 (79,2%) desses indivíduos que fumam ativamente registraram queixas relacionadas ao tabagismo, sendo que 34 (59,6%) deles receberam alguma forma de aconselhamento sobre o uso de tabaco. Ao todo, 9 (26,5%) indivíduos dentre os que receberam aconselhamento sobre tabaco e 1 (4,3%) que não teve aconselhamento registrado pararam de fumar (diferença de risco de 22,1%; IC 95%, [1,7%, 39,1%]; p=0,04). Dentre aqueles que receberam aconselhamento, 17 (50,0%) reduziram seus mpd, além de 7 (30,4%) daqueles que não tiveram aconselhamento (diferença de risco de 19,6%; IC 95% [-6,3%, 41,3%]; p=0,18). No geral, 14 (25,5%) dos 55 oftalmologistas que tiveram um paciente fumante ativo registraram evidências de aconselhamento sobre o uso de tabaco.
Conclusões: Os resultados deste estudo revelam tanto as oportunidades perdidas de aconselhamento sobre o uso do tabaco quanto a eficácia do aconselhamento no contexto de doenças oculares associadas à tireoide.
Keywords: Uso de tabaco; Aconselhamento; Doenças da glândula tireóide; Doença de Graves; Oftalmopatias
Arq. Bras. Oftalmol. 2024;87 (4 )
:1-8
| DOI: 10.5935/0004-2749.2021-0415
Abstract
Objetivo: Fenótipos Stargardt-like já foram associados a variantes patogênicas no gene ABCA4. O propósito desse estudo é descrever quatro pacientes com achados retinianos semelhantes a doença de Stargardt com resultados moleculares diferentes do esperado.
Métodos: Esse relato fez a revisão de prontuários médicos de quatro pacientes com distrofia macular e achados clínicos sugestivos de doença de Stargardt. Foram realizados avaliação oftalmológica, exames de imagens e testes usando next generation sequencing para avaliar variantes patogênicas associadas aos fenótipos dos pacientes.
Resultados: Os pacientes apresentavam atrofia macular e alterações pigmentares sugerindo achados clínicos de doença de Stargardt. Dois pacientes foram associados a genes com herança autossômica dominante (RIMS1 e CRX) e dois pacientes foram associados a genes com herança autossômica recessiva (CRB1 e RDH12) com variantes preditoras de serem patogênicas.
Conclusão: Distrofias maculares podem ter similaridades fenotípicas com fenótipo de Stargardt-like associados a outros genes além dos classicamente já descritos.
Keywords: Doença de Stargardt; Estudos de associação genética; Fenótipo; Padrões de herança; Sequenciamento de nucleotídeos em larga escala; Degeneração macular; Distrofias retinianas; Doenças genéticas
Arq. Bras. Oftalmol. 2025;88 (3 )
:1-8
| DOI: 10.5935/0004-2749.2024-0104
Abstract
PURPOSE: This study aimed to characterize retinitis pigmentosa associated with the eyes shut homolog gene, which causes hereditary retinal degeneration.
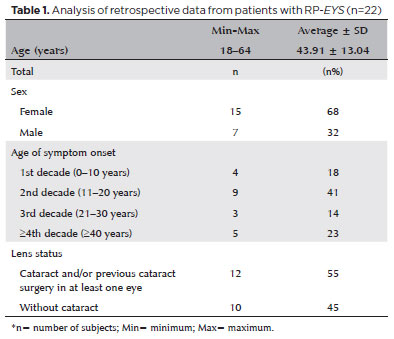
METHODS: The anatomical and functional findings of retinitis pigmentosa in patients with variants of the eyes shut homolog gene were characterized and compared using multimodal imaging and genetic analysis of the variants. Clinical data such as visual acuity, lens status, and refraction were obtained from medical records. Patients underwent an ophthalmic examination, including static visual field, microperimetry, optical coherence tomography, fundus autofluorescence, and fundus photography.
RESULTS: Twenty-two patients were included in the study. Several anatomical and functional characteristics of retinitis pigmentosa-eyes shut homolog were identified, including the presence of cataracts, cystoid macular edema, epiretinal membrane, and a tubular visual field. Genetic results revealed 26 distinct variants in the cohort, with 7 novel variants not previously documented or reported in the scientific literature or databases.
CONCLUSION: The findings demonstrate that eyes shut homolog-retinitis pigmentosa manifests in specific patterns, starting in adolescence with mild progression and advancing with age. The integration of multimodal imaging and genetic analysis has provided a detailed understanding of the anatomical and functional features of retinitis pigmentosa-eyes shut homolog. Seven novel variants of the eyes shut homolog gene have been identified. These findings enhance the understanding of eyes shut homolog-related retinitis pigmentosa characteristics of by detailing the spectrum of mutations in this gene within the Brazilian population.
Keywords: Retinal diseases/diagnostic imaging; Retinitis pigmentosa/genetics; Retinal degeneration; Eye proteins/genetics; Eye diseases, hereditary/genetics; Genes, recessive; Phenotype; Multimodal imaging; Tomography, optical coherence/methods; Fluorescein angiogr
Arq. Bras. Oftalmol. 2024;87 (2 )
:1-5
| DOI: 10.5935/0004-2749.2021-0395
Abstract
Objetivos: Avaliar a segurança e eficácia a longo prazo da vitreólise com Nd:YAG laser para moscas volantes sintomáticas, uma vez que permanece como um procedimento controverso devido a falta de evidência científica robusta sobre a manutenção dos resultados e ocorrência de efeitos adversos.
Métodos: Este estudo é uma extensão observacional de um ensaio clínico prospectivo, randomizado, duplo cego, previamente publicado. Oito de treze pacientes que foram submetidos a vitreólise com YAG laser foram acompanhados para uma reavaliação tardia, dezoito meses após o procedimento, para avaliar a eficácia e segurança do procedimento.
Resultados: Todos os pacientes mantiveram a melhora na sintomatologia notada ao final do procedimento original, com 25% dos casos apresentando melhora completa, e uma proporção semelhante (37,5%) demonstrando melhora significativa ou parcial. A melhora objetiva na opacidade foi similar ao achado no seguimento original de 6 meses. O questionário de qualidade de vida NEI-VFQ 25 não demonstrou diferença estatisticamente significativa nas respostas entre o sexto e o décimo oitavo mês de acompanhamento. Nenhum efeito adverso foi notado no exame clínico ou reportado pelos pacientes.
Conclusão: A eficácia da vitreólise observada ao sexto mês do acompanhamento foi mantida até o décimo oitavo mês, com todos os pacientes notando algum grau de melhora quando comparado ao estado pré procedimento. Nenhum efeito adverso tardio foi notado. Um ensaio clínico randomizado maior é necessário para confirmar a segurança do procedimento.
Keywords: Terapia a laser; Lasers de estado sólido; Vitrectomia; Corpo vítreo; Cirurgia vitreorretiniana; Acuidade visual; Doenças oculares; Qualidade de vida; Inquéritos e questionários
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
02-fig01.jpg)
06-tab01tb.jpg)
05-tab01tb.jpg)
02-fig01.jpg)
01-fig01.jpg)
14-fig01.jpg)