
Eduardo Ferrari Marback1; Milla Dias Sampaio2; Ricardo Danilo Chagas Oliveira3; Roberto Lorens Marback4
DOI: 10.1590/S0004-27492009000500025
RESUMO
Relatamos cinco casos de retinocitoma diagnósticados em um período de 75 meses. A média de idade ao diagnóstico foi de 193,2 meses. Dois casos apresentavam tumor unilateral, um bilateral e dois exibiam retinoblastoma no olho contralateral. As características clínicas e oftalmoscópicas, as implicações genéticas e a necessidade de tratamento e acompanhamento contínuo dos pacientes com retinocitoma são discutidas.
Descritores: Neoplasias da retina; Neoplasias da retina; Neoplasias da retina; Neoplasias da retina; Retinoblastoma; Relatos de casos
ABSTRACT
Five retinocytoma cases diagnosed over 75 months are reported. The mean age at diagnosis was 193.2 months. Two cases were unilateral, one was bilateral and two exhibited retinoblastoma in the fellow eye. Clinical and ophthalmoscopic aspects, genetical implications and the need of treatment and continuous follow-up of retinocytoma patients are discussed.
Keywords: Retinal neoplasms; Retinal neoplasms; Retinal neoplasms; Retinal neoplasms; Retinoblastoma; Case reports
INTRODUÇÃO
Retinocitoma é a variante benigna do retinoblastoma(1). É tumor de ocorrência rara, com potencial de malignização e mesmas implicações genéticas do retinoblastoma(2-5). A lesão pode representar um achado isolado, acompanhar retinoblastoma no mesmo olho ou olho contralateral, ou ser encontrada em familiares assintomáticos de portadores de retinoblastoma(2,5-6) .
Clinicamente o retinocitoma apresenta achados oftalmoscópicos similares aos padrões de regressão do retinoblastoma tratado por radioterapia: calcificação, aspecto em carne de peixe, alteração do epitélio pigmentar da retina (EPR) e atrofia coriorretiniana. A maioria dos tumores exibem duas ou mais destas características(1-3,5).
O objetivo deste trabalho é relatar nossa esperiência pessoal com 5 casos de retinocitoma (incluindo 2 previamente relatados)(6), mostrar suas características clínicas, comparando-as às do retinoblastoma, discutir suas implicações clínicas e genéticas, a necessidade de tratamento e acompanhamento.
RELATO DOS CASOS
No período de outubro de 2001 a janeiro de 2008, foram diagnosticados 5 casos de retinocitoma no Hospital Universitário Professor Edgard Santos, Universidade Federal da Bahia. Os dados demográficos destes pacientes encontram-se detalhados na tabela 1, resumidamente a média de idade ao diagnóstico foi de 193,2 meses (variando de 29 a 331 meses), sendo 2 casos unilaterais, 2 com retinoblatoma no olho contralateral e um com retinocitoma bilateral. No mesmo período, foram diagnosticados 62 novos casos de retinoblastoma, com média de idade ao diagnóstico de 36,9 meses (variando de 2 a 96 meses), sendo 46 unilaterais e 16 bilaterais.
Os detalhes do diagnóstico, número de tumores, sua localização, aspecto e conduta adotada foram os seguintes:
Caso 1
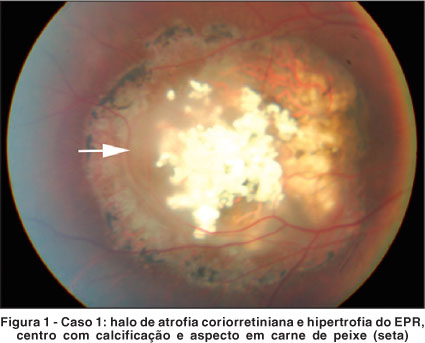
Tumor descoberto em mapeamento de retina para avaliar baixa de visão desde a infância. Lesão única na área macular do olho direito (OD) com halo de atrofia corioretiniana, hipertrofia do EPR e centro exibindo um misto de calficificação e aspecto em carne de peixe (Figura 1). Foi proposta fotocoagulação.
Caso 2
Paciente encaminhada por oncologista pediátrico após avaliação por leucocoria vista em fotos desde os 12 meses de vida. Tumor solitário em OD, envolvendo praticamente todo polo posterior. Halo de demarcação do EPR, centro exibindo calcificação e aspecto em carne de peixe (Figura 2). Realizada fotocoagulação.

Caso 3
OD enucleado por retinoblastoma aos 28 meses de vida. No olho esquerdo (OE), três tumores nasais ao disco óptico com aspecto em carne de peixe e lesão atrófica em meia periferia temporal. Inicialmente suspeitou-se de retinoblastoma e foi indicada quimioterapia. O paciente abandonou o acompanhamento, retornando após 21 meses, sem ter realizado qualquer tratamento. As lesões não apresentaram crescimento e exibiam trave vítrea entre as duas maiores. Iniciado tratamento por termoterapia transpupilar (TTT).
Caso 4
Compareceu solicitando correção estética para OE atrófico desde os 3 meses de vida. No OD havia tumor único, nasal e superior ao disco óptico, com halo tipo carne de peixe e calcificação central. Abandonou o acompanhamento após a enucleação do OE, que revelou retinoblastoma com regressão espontânea(6).
Caso 5
Pai de 2 irmãos com retinoblastoma bilateral. Foi convocado para exame, sendo identificados 3 tumores em cada olho. No OD havia dois tumores nas arcadas temporais, ambos com áreas de atrofia coriorretiniana, pontos de calficificação e centro tipo carne de peixe, uma terceira lesão nasal inferior ao disco tinha aspecto de atrofia coriorretiniana. No OE havia três lesões nasais ao disco óptico. Uma calcificada com halo tipo carne de peixe, uma menor com aspecto em carne de peixe e a terceira com atrofia coriorretiniana(6). O paciente abandonou o Serviço após ser informado do diagnóstico.
DISCUSSÃO
O diagnóstcio de retinocitoma carreia as mesmas implicações genéticas do retinoblastoma. Estima-se que 50% da prole de portadores de retinocitoma bilateral e/ou multifocal, e 10% da prole dos pacientes com lesão unilateral unifocal será afetada por retinoblastoma ou retinocitoma(2). É comum encontrar portador de retinocitoma como caso índice em famílias afetadas por retinoblastoma, como ocorreu no nosso caso 5. Por este motivo, todo paciente com diagnóstico de retinocitoma deve ser encaminhado para aconselhamento genético(2-3,5-6).
Como o retinocitoma é a variante benigna do retinoblastoma, compartilha com este sua origem genética. Para que o tumor se desenvolva é necessário que ocorra a inativação dos 2 alelos do gene RB1(5,7-8). A razão para a baixa frequência desse fenótipo nas grandes séries de retinoblastoma (1,8% a 10%) não é clara, mas estima-se que o retinocitoma seja subdiagnosticado(2,5). Em nosso Serviço, no mesmo período de tempo, diagnosticamos 5 casos de retinocitoma e 62 de retinoblastoma. Apenas o caso 3 apresentava retinoblastoma no olho contralateral e teve diagnóstico de retinocitoma aventado pela ausência de crescimento dos tumores não tratados, após abandonar o acompanhamento por quase dois anos.
A maioria dos pacientes com retinocitomas é assintomática, sendo o diagnóstico realizado usualmente por triagem dos parentes de pacientes com retinoblastoma, como no nosso caso 5, descoberto em exame de rotina, ou quando existe baixa de visão pela localização do tumor(2-3,5-6). O retinocitoma tipicamente não se apresenta com leucocoria, embora esta tenha sido vista no nosso caso 2(1,3,5). O tumor usualmente é diagnosticado mais tardiamente que o retinoblastoma. Apenas dois dos nossos casos eram crianças, o caso 3 com 29 meses e diagnóstico feito por apresentar leucocoria causada por retinoblastoma, comprovado por estudo anatomopatológico, no olho contralateral. Já o caso 2, único com leucocoria, tinha 54 meses ao diagnóstico, idade bem acima da média do retinoblastoma. Na série de Singh et al., a média de idade ao diagnóstico foi de 15 anos, similar a nossa que foi de 193,2 meses ou aproximadamente 16 anos(3).
O aspecto oftalmoscópico do retinocitoma mimetiza um ou, mais frequentemente, uma combinação dos padrões clássicos de regressão do retinoblastoma pós radioterapia(2-3,5). Em nossa série, os 5 pacientes exibiam um total de 13 retinocitomas. Destes, 7 tumores exibiam um padrão, 3 tumores a combinação de dois padrões, 2 tumores a combinação de três padrões e um tumor exibia a combinação de quatro padrões. Na série de Singh et al., 80% dos tumores apresentavam alguma combinação de padrões, enquanto 10% exibiam a combinação dos quatro padrões(3). Achado um pouco mais raro, e que não foi visto em nosso Serviço, foi a presença de semeadura do vítreo(3,9).
Duas teorias tentam explicar o motivo pelo qual a inativação dos alelos do gene RB1 resulta no surgimento do retinocitoma e não em retinoblastoma. A primeira propõe que nos casos de retinocitoma a inativação do segundo alelo do gene RB1 ocorreria num período mais tardio da maturação retiniana, quando a célula precursora afetada teria uma capacidade mitótica reduzida, resultando na formação de uma lesão com características citológicas benignas(5,10). A segunda hipótese é de que, em famílias com penetrância incompleta, a inativação do segundo alelo resultaria em produto parcialmente funcionante (a proteína que impede o surgimento do tumor)(5,11-12).
Histologicamente o retinocitoma apresenta população de células de tamanho e formato uniformes, citoplasma eosinofílico e abundante, formação frequente de "fleurettes" sem atividade mitótica ou necrose, com ocasionais focos de calcificação e hipertrofia do epitélio pigmentar da retina(1,5). Este aspecto histopatológico é completamente diverso daquele encontrado nos casos de regressão espontânea, como evidenciado pelo caso 4. Nestes últimos há gliose, fibrose e formação de células fossilizadas em olho atrófico(1,5-6).
O diagnóstico de retinocitoma implica na necessidade de acompanhamento continuado. Embora a transformação de retinocitoma em retinoblastoma seja rara, já foi documentada clinicamente e comprovada com base em achados histopatológicos(4). Alguns autores advogam inclusive, que os raros casos de retinoblastoma diagnosticados em crianças mais velhas e adultos, seriam na verdade resultado da transformação maligna de retinocitomas sem diagnóstcio prévio(5,13-14). Tal achado justifica o emprego de fotocoagulação ou TTT feito por nós, nas áreas com aspecto tipo carne de peixe em alguns dos nossos casos.
Em resumo, relatamos os achados em cinco casos de retinocitoma, destacando os aspectos oftalmoscópicos típicos. Trata-se de tumor subdiagnosticado, pois usualmente é assintomático. Pode estar associado ao retinoblastoma no mesmo olho ou no olho contralateral (como nos casos 3 e 4). Com relativa frequência, o retinocitoma ocorre em parentes de crianças com retinoblastoma (como no caso 5). Salientamos portanto a importância do exame de pais de pacientes com retinoblastoma, para melhorar o aconselhamento genético destas famílias, bem como a importância do acompanhamento de pacientes afetados por retinocitoma, ou mesmo o tratamento profilático das áreas com aspecto em carne de peixe, pois estas podem sofrer transformação maligna.
REFERÊNCIAS
1. Margo C, Hidayat A, Kopelman J, Zimmerman LE. Retinocytoma. A benign variant of retinoblastoma. Arch Opththalmol. 1983;101(10):1519-31.
2. Gallie BL, Ellsworth RM, Abramson DH, Phillips RA. Retinoma: Spontaneous regression of retinoblastoma or benign manifestation of the mutation? Br J Cancer. 1982;45(4):513-21.
3. Singh AD, Santos CM, Shields CL, Shields JA, Eagle RC Jr. Observations in 17 patiens with retinocytoma. Arch Ophthalmol. 2000;118(2):199-205.
4. Eagle RC Jr, Shields JA, Donoso L, Milner RS. Malignant transformation of spontaneous regressed retinoblastoma, retinoma/retinocytoma variant. Ophthalmology. 1989;96(9):1389-95.
5. Singh AD, Balmer A, Murnier F. Retinocytoma or retinoma. In: Singh AD, Damato B, Pe'er J, Murphree AL,Perry JD, eds. Clinical Ophthalmic Oncology. Philadelphia: Elsevier; 2007. p.487-90.
6. Marback RL, Sé DC, Santana Filho S, Fernandes Filho PM. Regressão espontânea de retinoblastoma. Arq Bras Oftalmol. 1982;45(5):133-7.
7. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68(4):820-3.
8. Aaby AA, Price RL, Zakov ZN. Spontaneous regressing retinoblastomas, retinoma, or retinoblastoma group 0. Am J Ophthalmol. 1983;96(3):315-20.
9. Lueder GT, Heon E, Gallie BL. Retinoma associated with vitreous seeding. Am J Ophthalmol. 1995;119(4):522-3.
10. Gallie BL, Dumm JM, Chan HS, Hamel PA, Phillips RA. The genetics of retinoblastoma: relevance to the patient. Pediatr Clin North Am. 1991;38(2):299-315.
11. Onadim Z, Hoog A, Baird PN, Cowell JK. Oncogenic point mutations in exon 20 of the RB1 gene in families showing incomplete penetrance and mild expression of the retinoblastoma phenotype. Proc Natl Acad Sci USA. 1992;89(13):6177-81.
12. Harbour JW. Molecular basis of low penetrance retinoblastoma. Arch Ophthalmol. 2001;119(11):1699-704.
13. Shields CL, Shields JA, Shah P. Retinoblastoma in older children. Ophthalmology. 1991;98(3):395-9.
14. Nork TM, Millecchia LL, de Venecia GB, Myers FL, Vogel KA. Immunocytochemical features of retinoblastoma in an adult. Arch Ophthalmol. 1996; 114(11):1402-6.
Endereço para correspondência:
Eduardo Marback
Rua Eduardo José dos Santos, 147 - Sl. 808
Salvador (BA) CEP 41940-455
E-mail: [email protected]
Recebido para publicação em 20.02.2009
Aprovação em 23.06.2009
Trabalho realizado no Hospital Universitário Professor Edgard Santos - Universidade Federal da Bahia - UFBA - Salvador (BA) - Brasil.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Martha Maria Motono Chojniak sobre a divulgação de seu nome como revisora, agradecemos sua participação neste processo.
© 2024 - All rights reserved - Conselho Brasileiro de Oftalmologia
![]()
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Scielo
Scielo
 Pocket
Pocket

{kind=link}